Protein structure determination by x-ray crystallography and NMR
In the last lectures we learned how protein structures are determined experimentally.
Today, we'll focus on how computational techniques are employed to aid
structure determination.
There are two main techniques for solving protein structures: x-ray
crystallography and Nuclear Magnetic Resonance (NMR).
As can be seen from the current
PDB holdings, more than 42,000 protein structures have been solved
so far, and are available from the
Protein Data Bank. About 13% of these are NMR structures, the rest
are x-ray crystallographic structures. As can also be seen on the PDB holdings list,
the number of solved proteins grows even faster, due to advancements
in structure determination techniques. Nevertheless, the number of
known protein sequences is almost an order of magnitude larger
(currently about 276,000, as available from the SWISS-PROT
database).
Reading PDB files
As we've learned last time, protein structures are stored at the Protein Data Bank, from which
individual protein structures can be retrieved as so-called PDB
files. Before we will turn to the structure determination itself, let
us have a closer look at a typical PDB file, to see what can be
learned about the background of the structure (experimental conditions
etc.) and the structural quality (the resolution, coordinate uncertainty).
We will focus on PDB entry 1TRO (an x-ray structure) as it can
be downloaded from the Protein Data bank.
The initial lines of a PDB entry contain information on the
protein, the source, the names of the people who sent the entry to the
PDB, references to literature that describe the structure in detail,
and some basic information regarding the crystallographic
data. Especially critical to check are the resolution and the R-factor
(or free-R factor), that contain information on how well the deposited
structure matches the measured data (x-ray reflection intensities in
this case).
HEADER DNA-BINDING REGULATORY PROTEIN 30-AUG-92 1TRO 1TRO 2
COMPND TRP REPRESSOR COMPLEX WITH OPERATOR 1TRO 3
SOURCE TRP REPRESSOR: (ESCHERICHIA COLI, STRAIN W3110); 1TRO 4
SOURCE 2 OPERATOR: SYNTHETIC 1TRO 5
AUTHOR Z.OTWINOWSKI,R.G.ZHANG,P.B.SIGLER 1TRO 6
REVDAT 1 31-JAN-94 1TRO 0 1TRO 7
REMARK 1 1TRO 8
REMARK 1 REFERENCE 1 1TRO 9
REMARK 1 AUTH Z.OTWINOWSKI,R.W.SCHEVITZ,R.-G.ZHANG,C.L.LAWSON, 1TRO 10
REMARK 1 AUTH 2 A.J.JOACHIMIAK,R.MARMORSTEIN,B.F.LUISI,P.B.SIGLER 1TRO 11
REMARK 1 TITL CRYSTAL STRUCTURE OF TRP REPRESSOR OPERATOR 1TRO 12
REMARK 1 TITL 2 COMPLEX AT ATOMIC RESOLUTION 1TRO 13
REMARK 1 REF NATURE V. 335 321 1988 1TRO 14
REMARK 1 REFN ASTM NATUAS UK ISSN 0028-0836 006 1TRO 15
[..]
REMARK 2 1TRO 23
REMARK 2 RESOLUTION. 1.9 ANGSTROMS. 1TRO 24
REMARK 3 REFINEMENT. 1TRO 26
REMARK 3 PROGRAM PROLSQ 1TRO 27
REMARK 3 AUTHORS KONNERT,HENDRICKSON 1TRO 28
REMARK 3 R VALUE 0.167 1TRO 29
REMARK 3 RMSD BOND DISTANCES 0.015 ANGSTROMS 1TRO 30
REMARK 3 RMSD BOND ANGLES 1.3 DEGREES 1TRO 31
Following the introductory material, some specific information regarding the protein and its crystalline form are provided, including the protein sequence, secondary structure, disulfide bonds (where present) and the like. Also the dimensions of the unit cell, the space group and related information are given.
SEQRES 1 G 108 MET ALA GLN GLN SER PRO TYR SER ALA ALA MET ALA GLU 1TRO 106
SEQRES 2 G 108 GLU ARG HIS GLN GLU TRP LEU ARG PHE VAL ASP LEU LEU 1TRO 107
SEQRES 3 G 108 LYS ASN ALA TYR GLN ASN ASP LEU HIS LEU PRO LEU LEU 1TRO 108
[..]
FORMUL 13 HOH *572(H2 O1) 1TRO 124
HELIX 1 1A TYR A 7 GLN A 31 1 1TRO 125
HELIX 2 1B HIS A 35 MET A 42 1 1TRO 126
HELIX 3 1C ASP A 46 ARG A 63 1 1TRO 127
[..]
HELIX 23 4E ILE G 79 ALA G 91 1 1TRO 147
HELIX 24 4F VAL G 94 LEU G 105 1 1TRO 148
CRYST1 43.650 72.430 107.390 90.00 94.96 90.00 P 21 8 1TRO 149
Then come the actual atom coordinates (or structure), whose listing
takes up most of the average PDB file. Each listing begins with
"ATOM" and is followed by:
- The atom number
- The atom name (which is uniquely defined for each type of amino acid)
- The type of residue
- The chain identifier: In this case "A" indicating that this is
one of several molecules in the model.
- The residue's sequence number in the protein
- The x, y and z coordinates
- The occupancy (the fraction of atoms in the
crystal that occupies the specified coordinates, usually one).
- The temperature factor (or B factor), a measure for the coordinate uncertainty.Low B-factors (< 30) therefore correspond to well-defined parts of the structure, whereas high B-factors (> 80) might indicate highly disordered parts of the structure or even mis-interpreted parts of the model.
- And lastly, the models identifier (1TRO in this case) and the line number in the file).
ATOM 1 N SER A 5 4.462 27.337 23.235 1.00 61.04 1TRO 156
ATOM 2 CA SER A 5 5.077 28.626 22.900 1.00 62.14 1TRO 157
ATOM 3 C SER A 5 4.851 28.971 21.434 1.00 63.65 1TRO 158
ATOM 4 O SER A 5 4.235 28.222 20.647 1.00 65.52 1TRO 159
ATOM 5 CB SER A 5 6.545 28.668 23.270 1.00 58.51 1TRO 160
ATOM 6 OG SER A 5 7.320 27.704 22.589 1.00 50.29 1TRO 161
This goes on for a while, until the end of the peptide chain, which is marked by the "TER" line. If there are any other molecules that co-crystallized with the protein (such as solvent molecules or ligands) they are listed as "hetero-atoms" near the end of the file.
TER 4039 C J 19 1TRO4194
HETATM 4814 O HOH 1 20.236 5.712 14.585 1.00 49.26 1TRO4969
HETATM 4815 O HOH 2 30.581 1.151 18.292 1.00 29.71 1TRO4970
HETATM 4816 O HOH 3 28.781 -11.725 8.943 1.00 51.99 1TRO4971
HETATM 4817 O HOH 4 9.273 -10.253 29.757 1.00 56.19 1TRO4972
x-ray crystallography

The main technique for determining protein structures is x-ray
crystallography. Since the first protein structure (myoglobin) was solved by this
technique by John Kendrew and Max Perutz in the late fifties, several
thousand others followed. As can be appreciated from the picture on the right,
which shows John Kendrew with the structural model of myoglobin, at
that time the determination of a structure the size of a protein,
without the aid of a computer, was a formidable task.
It is important to note that in both x-ray crystallography and NMR,
protein structures are not measured directly in the
experiment. Rather, a set of data is collected (a diffraction pattern
or a NMR spectrum), from which a model of the protein structure
is derived. To appreciate the difference between data and structure,
we'll now look at two different structures of the same protein, and
the corresponding x-ray crystallographic data. For this, we will
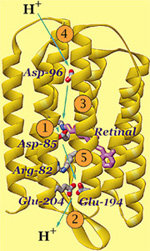
concentrate on the bacterial light driven proton pump
bacteriorhodopsin. Click
here for more background information on bR.
First download two bR structures from the Protein Data Bank, with PDB entries
1BRR and 1QHJ. Save both PDB files to your local account (see the last
lecture if you forgot how to download from the PDB).
View the structures with rasmol:
rasmol 1BRR.pdb
to focus only on one of the three protein molecules in the file type
(in rasmol):
restrict *a
to see the main features of the structure:
cartoons
color structure
To highlight the dye (the light sensor in the protein interior):
select ret and *a
cpk
Have a close look at the structure, before repeating the procedure for 1QHJ.pdb
in a different window, such that you can compare the two structures.
Question:
By looking at the structures, which of the two structures would you
prefer, in terms of coordinate accuracy?
Remember, so far we only looked at the coordinates, which represent a model
that was optimized against the actually measured data. So, let
us now have a look at the data. In x-ray crystallography, data are
collected by measuring a diffraction pattern that is obtained from
x-rays reflected by a protein crystal. As mentioned in the lecture,
this diffraction pattern itself does not suffice to determine the complete structure
since only the amplitudes of the diffracted waves were collected, not
their phases. In x-ray crystallography, however, there are a number of
tricks available (e.g. isomorphous replacement, molecular replacement)
but we will not go into that in detail here. What is important to
remember is that eventually, an atomic electron density map is obtained.
Question:
Why do primarily the electrons of a molecular sample contribute to the
diffraction of x-rays? answer.
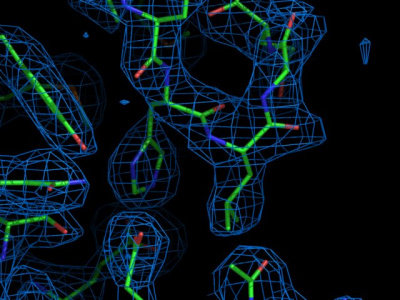
Visit the Uppsala electron density server
to view the electron density map 1BRR. Enter the PDB code (1BRR), and
wait for the summary page to load. Several plots with information on
this structure are available. Feel free to browse around to check the
meaning of the individual plots. At the bottom of the summary, the
electron density viewer can be activated. Select the "Upssala viewer"
instead of the default "Astex viewer" and input that the map should be
centered around residue A83 before clicking "Go". After a while, a
java applet should appear with the electron density and the model
structure visible. Shift the map somewhat to focus on the side-chain
of residue Tyr 83 (by clicking on "center" and then in the atom that
should be centered, until you are in the six-membered aromatic ring).
Do you find the electron density for the aromatic
ring convincing? Now shift the focus to residue A35 (by entering "A35"
in the text field under "center" and then hitting "center"). How is
the fit between the model and the data here? To see the retinal, the
light sensor in the center of the protein, center on A999.
Now repeat the procedure for entry 1QHJ. How is the fit for residue
A35? and for A83? And the retinal (called A300 here)?
Question:
Based on the data and on the model structures, would you say there is
a large impact of the resolution of the data on the accuracy of the structural model?
The highest resolution x-ray crystallographic structures have a
resolution of approx. 0.8A or even somewhat better. To see an example
of such a dataset, look at the density for structure 1G6X, and center
around residue A23. Do you recognize the difference in appearance of
the Tyrosine ring?
Question:
Although the resolution of this structure is rather high, at 0.86A, the
hydrogen atoms (e.g. on the side chain of TYR23) are hardly
visible. Why is this? answer.
NMR
The other main technique for determining protein structures is NMR. In
contrast to x-ray crystallography, no crystals are required for an NMR
experiment. Rather, the structure is determined of the protein in
solution. Therefore, it has the advantage that the protein can be
studied in its native environment. On the other hand, the resolution
of an NMR structure is usually lower and there is a size limitation of
a few hundred amino acids for structure determination using NMR.
It would go beyond the scope of this course to explain the
NMR experiment in detail. We will therefore only briefly touch on the
experimental setup and then focus on the structure building and
refinement step based on the obtained data.
The NMR signal is recorded as a nuclear magnetic resonance spectrum of predominantly the
hydrogen atoms after the sample has been subjected to a (number of)
strong magnetic pulse(s). Mainly hydrogen atoms give rise to the
signal, because of the magnetic spin properties of the hydrogen
nucleus (a proton). The naturally occurring isotopes of the other
elements that are found in proteins, carbon (12C) and
oxygen (16O),
have a zero nuclear magnetic moment. Nitrogen (14N) does
have a non-zero magnetic moment, but can usually not be used in NMR,
for reasons that would go beyond the scope of this course to explain.
These elements, therefore, can only
be utilized in NMR experiments when chemically replaced by a specific
isotope, like 13C or 15N. The most structurally
relevant information is usually obtained from a so called NOESY
experiment (Nuclear Overhauser Enhancement SpectroscopY).
The Nuclear Overhauser Effect or Nuclear Overhauser Enhancement is the
change (enhancement) of the signal intensity from a given nucleus as a
result of exciting or saturating the resonance frequency of another
nucleus. Since this effect is distance-dependent, it can be used to
derive the distance between an interacting pair of protons. In
practice, protons closer than 6A apart can be identified this way.
Now, we will calculate a model of the structure of a small protein,
the B1 domain of protein G,
from the proton-proton distance information obtained from a NOESY
experiment. Download the data file containing the distance information
here. You can have a look at the file
(with the program "more" or "less" or a browser or editor of your
choice) to assure yourself that there are indeed only distance bounds
listed in this file. Additionally, we need an initial guess of the
structure. Since we don't know the structure yet, we have to start from
an unstructured peptide chain, which can be obtained here . Have a look at the structure with:
rasmol proteinG.pdb
Finally, we need something called a
molecular topology, a chemical description of the protein: which atoms
does the molecule contain, which atoms are covalently bonded to each
other, etc. This molecular topology file is available here . Now, in principle, we have all the data
to attempt to build a structure that is in agreement with all
experimentally determined distances. The only thing we still need is
an input file for the
CNS program.
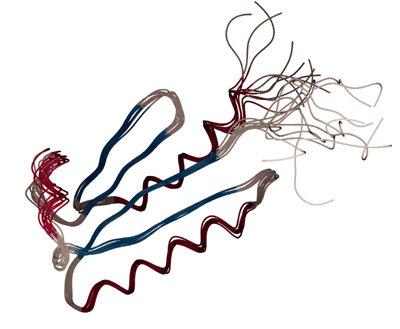
Note that, in contrast to x-ray crystallography, where a
single structure is presented, to reflect the fact that the NMR experiment probes
an ensemble of protein molecules in solution, an NMR structure is
usually represented by an ensemble of structures, that all fulfill the
NMR data. Start CNS with
tcsh
source /usr/global/cns/cns_solve_env
cns < anneal.inp
10 structures of the B1 domain of protein G will now be calculated by
simulated annealing. This is a computationally intensive calculation,
as the structure is slowly, dynamically transformed from the extended
starting conformation to the real structure, by a slow-cooling
simulation, also called simulated annealing. As the calculation is
running, we can have a look at how exactly such a calculation proceeds,
and how the final structure is generated from the starting
guess. Download this file, open another shell, and
open the just downloaded file in pymol:
/usr/global/whatif/pymol/pymol sa.pdb
on the white bar, in the top pymol window, type:
show cartoon
and then press the "play" button at the bottom right of the main pymol
window, to see an animation of the simulated annealing structure
calculation procedure. If the movie plays too fast, on the menu, under
movie -> speed, choose a different speed.
When the CNS structure calculation has finished, switch back to that
window and type:
cat anneala_*.pdb > anneal.pdb
to combine all ten generated structures into one file. View the result
with:
rasmol anneal.pdb
Question:
Which parts of the structure are well-defined, and which parts show
more ambiguity?
There is also a x-ray structure available of the B1 domain of protein
G, available under the PDB code 1PGB. Download it from the Protein Data Bank and compare it to
the just calculated NMR structure.
Question:
What are the main differences between the NMR and x-ray structures of
the B1 domain of protein G? hint