Simulation of Atomic Force Microscope Stretching Experiments II - Mechanical Properties of Polyethylenglycol (PEG)
Berthold Heymann and Helmut Grubmüller
Collaborators: Filipp Oesterhelt and Hermann Gaub (LMU Munich), Support: DFG (SFB 143/C1)
Unfolding and elastic properties of single poly(ethylene-glycol) (PEG) polymers were studied by means of molecular dynamics (MD) simulations. The simulations were performed in close resemblance to recent single molecule force spectroscopy experiments that were carried out in Hermann Gaubs lab in Munich (Germany), and this allowed an interpretation of these experiments at the atomic level.
 and PEG in water (bottom).")

To study the influence of the solvent on the unfolding and elastic properties of PEG, two simulation models of PEG, consisting of 18 monomers, were used: PEG in gas phase (Figure 1, top), which is intended also to describe PEG in very apolar solvents, and PEG in water solvent (Figure 1, bottom). For both, extended stretching simulations were carried out independently. Force extension curves for gas phase PEG and solvated PEG were extracted from the simulations (see Figure 2).
 and 7.5 m/s (bold line), respectively, as well as for PEG in gas phase (thin line). Three force regimes (I, II, and III) are distinguished and discussed in the text.")
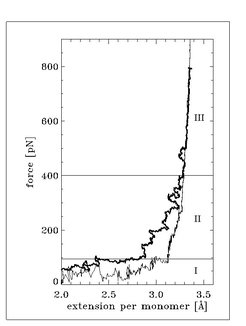
Force extension curves for gas phase PEG and solvated PEG in water extracted from the simulations are shown in Figure 2. We distinguish three regimes: the first regime (I) up to stretching forces of about 100 pN, where the elasticities of the polymers (which is proportional to the slope of the respective force extension curves) do not exhibit significant differences; the second regime (II) in the range between 100 and 400 pN, where the force extension curves significantly deviate from each other; and the third regime (III) above 400 pN, in which the force extension curves are nearly identical.
What is the reason for the deviation in the second regime at stretching forces between 100 and 400 pN?
To answer this question, we look at what happens at the atomic level during stretching. As visible from the first snapshot (see Figure 3), the hydrated PEG assumes a helical structure, what is not the case for gas phase PEG. This helical structure is stabilized by water molecules that form hydrogen bonds to up to four neighboured monomers simultaneously. These intermonomeric interactions, mediated by water molecules, are called water bridges.

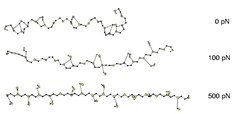
When the polymer is stretched, the distance between the monomers is increased. For stretching forces above 100 pN the distances are so large that only water bridges between adjacent monomers can be formed (second snapshot). At a stretching forces of 500 pN (third snapshot) the polymer is nearly linear, and even interactions between adjacent monomers cannot be mediated by water molecules.

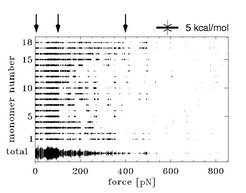
This finding is illustrated in Figure 4. Here, the strength of the water bridges between adjacent monomers is shown as a function of the stretching force. The strength is indicated by the thickness of the 18 traces. The arrows on the top mark the beginning of the respective regimes in the force extionsioncurves. It is visible that the strength of the water bridges significantly decreases in the second regime, in which the helical structure completely disappears, and that almost no water bridges are present in the third regime.