
Research
Overview and summary
Dynamics at surfaces is the field where atomic-scale understanding emerges concerning the elementary events important to interfacial energy transfer and surface chemistry. Following the tradition of gas-phase chemical dynamics, a well-proven strategy is employed, where quantum-state, angle and speed resolved experiments are compared with first-principle theoretical simulations.
Theoretical understanding of surface chemistry will eventually become a tool to design new chemical technology including heterogeneous (photo) catalysts, photovoltaics, fuel cells and much more. To reach this goal, we require new ideas and new theories of molecular interactions at interfaces. Applying cutting-edge laser, molecular beam, and ultrahigh vacuum technology to design well-defined experiments that can catch molecules in the act of reacting, our group strives to provide benchmark measurements that set standards for the next generation of theoretical advance. In particular, we seek to discover the "rules" that govern the conversion of energy at interfaces. Although too small to see with the naked eye and too fast to follow except with the fastest pulsed lasers, energy conversion takes place one molecule at a time and one collision at a time. By isolating these individual energy conversion events and studying them, we are building the conceptual bridge connecting our macroscopic experience of energy conversion to the molecular world.
Research concepts and strategies


In defining research concepts and strategies it is helpful to first ask the question: what distinguishes dynamics at surfaces from the well-studied field of dynamics in gas-phase chemical reactions. Many points come immediately to mind. First, the interactions present at a solid interface may dramatically alter the potential energy surface (PES), for example raising or lowering barriers to reactions in comparison to their gas phase counterparts. This is the principle of heterogeneous catalysis. The theoretical methods for calculating these changes in the PES are popular but not well tested. A second essential aspect of dynamics at surfaces concerns the fact that, in contrast to gas phase phenomena, the solid is an energy bath that can influence the outcome of chemical processes by supplying or draining energy from the reaction center in ways that are not possible in the gas phase. How translational energy is taken up by the solid during a molecular collision can influence the molecular trapping probability, the first step in all catalysis. How energy in molecular vibration exchanges with the solid can control the energy directly available to make and break chemical bonds. The research strategies used in the Department of Dynamics at Surfaces are designed to gain quantitative information about both of these essential defining aspects of the field.
State-to-state molecular-beam surface-scattering


Nd:YAG pumped dyelaser used for preparation and detection of molecules.
In order to observe how a molecule exchanges energy with a surface we perform scattering experiments in ultra-high vacuum conditions. In such experiments, the molecule is first prepared in a specific initial state prior to the collision with the surface. Here, the speed, direction, degree of orientation, rotational, vibrational and electronic motion are all defined. After the collision, all these quantities are re-measured. Using theoretical methods we can determine what happened to the molecule during the collision.
Combining molecular beams with laser-based optical pumping methods, one can precisely control the speed, direction and quantum state of the molecule. Using quantum-state selective detection, for example Resonant Enhanced Multiphoton Ionization (REMPI), the quantum state of the molecule after the collision is determined. Using pulsed lasers separated over a distance, one may perform state-to-state, time-of-flight (TOF) measurements, which reveal the speed and direction of the recoiling molecule.
Velocity resolved kinetics
Catalysts are widely used to increase reaction rates. They function by stabilizing the transition state of the reaction at their active site, where the atomic arrangement ensures favorable interactions. However, mechanistic understanding is often limited when catalysts possess multiple active sites-such as sites associated with either the step edges or the close-packed terraces of inorganic nanoparticles with distinct activities that cannot be measured simultaneously. Using molecular beams to controllably introduce reactants and slice ion imaging to map the velocity vectors of the product molecules, which reflect the symmetry and the orientation of the active site, we are able to measure the reaction rates at different active sites simultaneously.

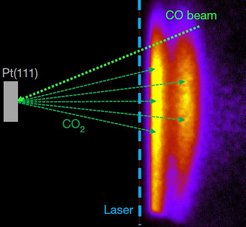
An example is the oxidation of carbon monoxide over platinum surfaces, one of the oldest and best studied heterogeneous reactions. We used our velocity-resolved kinetics approach to map the oxidation rates of carbon monoxide at step edges and terrace sites on platinum surfaces, and find that the reaction proceeds through two distinct channels: it is dominated at low temperatures by the more active step sites, and at high temperatures by the more abundant terrace sites. We expect our approach to be applicable to a wide range of heterogeneous reactions and to provide improved mechanistic understanding of the contribution of different active sites, which should be useful in the design of improved catalysts.
Surface scattering at extreme ultra-violet (SUV) free electron lasers
The “Mobile Beamer” is designed to make use of the advantageous properties of the radiation produced by the XUV free electron laser in Dalian (DCLS, China) for surface scattering experiments on atoms and molecules. The XUV photolysis of small molecules will be used as a novel source of atomic beams, providing fine control over the electronic state and kinetic energy. In addition, the tunable XUV light source will be used for sensitive, isomer-specific detection of molecular scattering products and reaction intermediates at surfaces, via photoionization mass spectrometry with high temporal time of flight and spatial velocity map imaging resolution.
 crystal at 830°C held in an ultra-high vacuum chamber")

With this powerful experimental tool in hand we would like to answer fundamental questions in the field of surface chemistry. For instance, can electronic excitation of incident atoms lead to an increased reactivity with surface adsorbates or is the electronic energy lost to surface degrees of freedom without enhancing a reaction? To answer this and other questions a German-Chinese joint research group was founded benefitting from the expertise of Prof. Xueming Yang’s workgroup on spectroscopy, gas phase reaction dynamics, as well as the free electron laser and the broad knowledge on building and designing surface scattering experiments available in our group in Göttingen.
Ultra-Short H-atom pulses
Ultra-short light pulses have become invaluable in time-resolved studies in chemistry and physics and processes that occur on a sub-picosecond time-scale can be resolved. However, many important processes are initiated by collisions. While lasers have revolutionized experiments using light pulses, experimentally proven concepts for producing ultra-short pulses of neutral matter are still in their infancy.
In our experiments, we use ultra-fast lasers for photolysis and bunch-compression photolysis, a technique developed in our department recently [1], to generate ultra-short H-atom pulses. In a proof-of-principle experiment we already produced H-atom pulsed with 1.2 ns duration. The method applies an ultra-short femtosecond laser pulse for photolysis of jet-cooled HI. The wavelengths of the broadband laser pulses are spatially ordered in a way that the H-atom pulse compresses after a defined distance due to the large spread in velocity. The usage of a spatial chirp increases the photolysis volume and therefore the method generates high intensity H-atom pulses for short pulse durations.
We are now improving the bunch-compression photolysis method using shorter photolysis wavelengths and more intense laser pulses with the aim of studying the interaction of H-atoms with photo-excited surfaces on a sub-nanosecond time-scale.
First principles simulations of molecule-surface dynamics
To augment the main effort of our lab, which is experimental in nature, we also carry out forefront research in theoretical modeling of surface chemistry and dynamics. Density functional theory, quantum dynamics, and mixed quantum classical methods are all key approaches that allow us to reproduce experimental observation from first principles in a computer simulation. With the most highly developed theories that faithfully reproduce a variety of experimental observations, we may explore molecular dynamics at surfaces in much greater atomic level detail through the methods of computer simulation.