Epigenetik: Regulation der Genaktivität durch Histonmodifizierungen
Forschungsbericht (importiert) 2008 - Max-Planck-Institut für Multidisziplinäre Naturwissenschaften
Chromatin
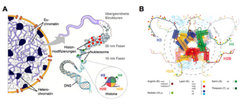
Die DNS als Träger der Erbinformation liegt in den Kernen aller Zellen unseres Körpers in einem Komplex mit basischen Proteinen als sogenanntes Chromatin vor. Dabei sind kurze Abschnitte DNS jeweils um ein Set von Histonproteinen (H2A, H2B, H3 und H4) gewickelt. Diese als Nukleosom bezeichnete Anordnung stellt die fundamentale, sich wiederholende Einheit des Chromatins dar (siehe Abb. 1A). Durch Aneinanderreihung der Nukleosome entsteht eine sogenannte Perlenkettenstruktur (10 nm-Faser). Es wird angenommen, dass diese wiederum spiralförmig zu Solenoidfasern (30 nm-Faser) organisiert vorliegt. Allerdings sind diese sowie weitere übergeordnete Faltungsprinzipien der Chromatinstruktur nach wie vor nicht vollständig geklärt.
Durch Anfärben der DNS mit basischen Farbstoffen können lichtmikroskopisch sowie ohne spezielle Behandlung direkt im Elektronenmikroskop Bereiche unterschiedlich dichten Chromatins differenziert werden. Dabei stellt das weniger dichte Euchromatin den Anteil des überwiegend transkriptionell aktiven Chromatins dar: Es enthält die Mehrheit der Gene, hat nur wenige sich wiederholende DNS-Abschnitte und kann in der Meiose frei rekombiniert werden. Das verdichtete Heterochromatin enthält demgegenüber nur sehr wenige Protein-kodierende DNS-Abschnitte. Es wird überwiegend aus sich stark wiederholenden Sequenzabschnitten der DNS gebildet. Außerdem findet in diesen Bereichen nur selten meiotische Rekombination statt. Obwohl klar ist, dass sich Eu- und Heterochromatin nicht nur auf physiologischer, sondern auch auf biochemischer Ebene unterscheiden, ist der molekulare Aufbau der unterschiedlichen Chromatinarten bisher unbekannt.
Histonmodifizierungen und Chromatindomänen
Während die Nukleosomen des gesamten Chromatins auf die gleiche Weise zusammengebaut sind, können sich molekulare Unterschiede zwischen verschiedenen Nukleosomen vor allem durch zahlreiche chemische Veränderungen der Histonproteine ergeben (sogenannte post-translationale Modifizierungen). Diese scheinen als Signale oder Marker zu fungieren, die es der Zelle erlauben, unterschiedliche Regionen des Chromatins zu erkennen und zu definieren. Die Wissenschaftler nehmen an, dass post-translationale Modifizierungen der Histonproteine essentiell sind für Regulationsmechanismen, die die Auslesung der Erbinformation in allen unseren Zellen steuern.
Alle vier Histone, die am Aufbau eines Nukleosoms beteiligt sind, können auf vielfältige Art post-translational modifiziert werden. Arginin- und Lysinreste können in definiertem Ausmaß methyliert werden (für Details siehe Abb. 1B). Lysinreste können darüber hinaus acetyliert werden. Auch wurde das Anheften der regulatorischen Polypeptide Ubiquitin und SUMO (ein Ubiquitin-ähnliches Protein) an Lysine beschrieben. Zahlreiche Serin- und Threoninreste sowie wenige Histidinreste können phosphoryliert werden. Wie Abbildung 1B zeigt, ist die Anzahl der möglichen Histonmodifizierungen, durch die ein Nukleosom indiziert werden kann, immens. Allerdings ist bisher unklar, ob alle Markierungen unabhängig voneinander und gleichzeitig vorkommen können. In der Tat gibt es Hinweise, dass die Modifizierung bestimmter Reste die Markierung anderer Aminosäuren in den Histonen beeinflusst [1]. Außerdem können zahlreiche Lysinreste auf mehrere Arten modifiziert werden (zum Beispiel Methylierung oder Acetylierung; Acetylierung oder Ubiquitinierung). Die meisten Histonmodifizierungen werden auf den aminoterminalen Schwanzregionen der Histone gefunden. Diese ragen aus dem Nukleosom heraus und können mit anderen Faktoren wechselwirken. Obwohl Histonmodifizierungen schon vor über 40 Jahren zum ersten Mal beschrieben wurden, konnten Enzymsysteme, die die post-translationale Modifizierung der Histone steuern, erst durch intensive Arbeiten in den letzten zehn Jahren identifiziert und charakterisiert werden.
 Hierarchischer Aufbau des Chromatins in einer eukaryontischen Zelle (Interphase). Das Nukleosom stellt die fundamentale, sich wiederholende Einheit des Chromatins dar. Es besteht aus einem Proteinkomplex der vier Histone (H2A, H2B, H3 und H4), umwickelt von 170-200 bp (Basenpaaren) an DNS. (B)Schematische Darstellung eines Nukleosoms. Sogenannte aminoterminale Schwanzregionen der Histone ragen aus dem Nukleosom heraus. Unterschiedliche Arten und Positionen post-translationaler Histonmodifizierungen sind markiert. me, Methylierung; ac, Acetylierung; su, SUMOylierung; ub, Ubiquitinierung; ph, Phosphorylierung. Die gestrichelte Linie entspricht der Position der DNS.")
Die Herstellung spezifischer Antikörper, die bestimmte Histonmodifizierungen in definierter Sequenzumgebung erkennen, hat es in den vergangenen Jahren ermöglicht, die Zellbiologie unterschiedlicher Histonmarkierungen zu untersuchen. Dabei wurden bestimmte Histonmodifizierungen in definierten Bereichen des Chromatins gefunden (Abb. 2). Außerdem konnten die Wissenschaftler zeigen, dass auch die verschiedenen Methylierungsstufen ein- und desselben Lysinrestes eine ganz unterschiedliche Verteilung aufweisen. Diese Beobachtungen deuten darauf hin, dass verschiedene Muster an Histonmodifizierungen unterschiedliche Bereiche des Chromatins markieren [2]. Es wird angenommen, dass Histonmodifizierungen kausal an der Entstehung solcher Chromatindomänen beteiligt sind. Während die molekularen Zusammenhänge bisher weitgehend ungeklärt sind, konnten in der Tat nicht überlappende Muster an Histonmodifizierungen eu- und heterochromatischen Chromatindomänen zugewiesen werden.
Histonmodifizierungen und Epigenetik
Neben der Zuordnung unterschiedlicher Histonmodifizierungen zu verschiedenen Bereichen/Domänen des Chromatins und damit des Genoms werden vor allem die biologischen Funktionen der Histonmodifizierungen intensiv untersucht. Methylierungen von Lysinresten der unterschiedlichen Histone scheinen hier von besonderer Bedeutung. So konnte gezeigt werden, dass Methylierungen verschiedener Lysinreste die lokale und globale Genaktivierung und -inaktivierung steuern. Dabei nehmen die Lysinmethylierungen Einfluss auf alle Ebenen der transkriptionellen Regulation (Zugang des Transkriptionsapparates, Start der Transkription, Elongation der Transkription, Reinitiierung der Transkription, etc.). Da Methylierungen im Gegensatz zu den meisten anderen post-translationalen Histonmodifizierungen biochemisch besonders stabil sind, wird ihnen ein besonderes Indizierungspotential zugeschrieben.
 Anfärbung polytener Chromosomen der Fruchtfliege Drosophila mit Antikörpern (α-Histon PTM a, b), die unterschiedliche Histonmodifizierungen erkennen (rot). Gesamt-DNS wurde mit dem Farbstoff DAPI eingefärbt (blau). Die Pfeile markieren Regionen der sog, Zentromere, die Bereiche besonders stark verdichteten Chromatins darstellen. (B) Anfärbung von Fibroblastenzellen der Maus mit Antikörpern, die denselben Lysinrest in Histon H3 erkennen, allerdings in unterschiedlichen Methylierungsstufen (mono- (me1), di- (me2) und tri- (me3) methyliert). Gesamt-DNS wurde mit dem Farbstoff DAPI eingefärbt (blau). Die unterschiedlichen Histonmodifizierungen lassen sich verschiedenen Bereichen des Chromatins zuordnen (Schemata jeweils auf der linken Seite).")

Nach der Entzifferung des menschlichen Genoms ist klar, dass neben der Variation der Gene der variablen Kontrolle der Auslesung der Erbinformation eine wesentliche Bedeutung bei der Entstehung von Phänotypen zukommt. Tatsächlich wurde bereits vor etlichen Jahren erkannt, dass bestimmte vererbbare biologische Merkmale nicht durch unterschiedliche Allele, sondern durch unterschiedliche Aktivität ein und desselben Allels hervorgerufen werden. Diese sogenannten epigenetischen Effekte (= vererbbare Kontrolle der Genauslesung) scheinen auf der Ebene des Chromatins unter anderem über stabile Histonmodifizierungsmuster (insbesondere Methylierungen) vermittelt zu werden. Eine dauerhafte, von Zelle zu Zelle vererbte Regelung der Genaktivität spielt zum Beispiel in der Zelldifferenzierung und embryonalen Musterbildung, aber auch bei der krebsartigen Veränderung von Zellen eine bedeutende Rolle.
Wie funktionieren Histonmodifizierungen auf molekularer Ebene?
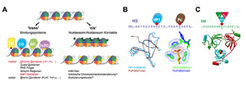
Um die Bedeutung der unterschiedlichen Histonmethylierungsstellen und -formen zu verstehen, ist es nötig, die molekularen Mechanismen zu analysieren, über die diese Signale in biologische Funktionen umgesetzt werden. Generell können Histonmodifizierungen entweder direkt das Aneinanderreihen der Nukleosome (also deren Packung) beeinflussen, indem sie die Interaktion benachbarter Nukleosome steuern (Abb. 3A). Alternativ können Histonmodifizierungen aber auch spezifisch die Wechselwirkung der Histone mit Bindungsproteinen regulieren. Die Wissenschaftler nehmen an, dass derartige Bindungsproteine die Chromatinstruktur und -dynamik steuern.
Die Arbeiten der Forscher am Göttinger Max-Planck-Institut haben zur Identifizierung und Charakterisierung mehrerer spezifischer Bindungsproteine für definierte Histonmethylierungen beigetragen. So konnte gezeigt werden, dass sogenannte Chromo-Domänen in den Heterochromatinprotein 1 (HP1)- und Polycomb (Pc)-Faktoren die Histon H3-Lysinmethylierungen an Position 9 (H3K9me) und Position 27 (H3K27me) spezifisch erkennen [3]. Während diese Proteine vor allem die Di- und Trimethylierungsstufen der entsprechenden Lysinreste binden, konnte des Weiteren gezeigt werden, dass Faktoren mit sogenannten MBT-Domänen (zum Beispiel L3MBTL1) nur mit mono- und dimethylierten Lysinresten der Histone wechselwirken [4, 5]. Strukturelle und biophysikalische Untersuchungen haben zu einem relativ genauen Verständnis der Interaktion dieser Bindungsproteine mit methylierten Histonen geführt (Abb. 3B, C) [3, 5, 6].
Während in den letzten Jahren einige weitere Bindungsproteine für Histonmodifizierungen und vor allem für spezifische Lysinmethylierungen gefunden wurden, konnten die Göttinger Wissenschaftler elementare Prinzipien der Rekrutierung dieser Faktoren entschlüsseln. Sie zeigten, dass HP1-Bindung an H3K9me dynamisch über eine benachbarte Phosphorylierung des Serinrestes an Position 10 (H3S10ph) reguliert wird [7]. Außerdem konnten physiologische Zusammenhänge zwischen der Rekrutierung von Bindungsproteinen und der Entstehung und Aufrechterhaltung von Chromatindomänen aufgedeckt werden [8, 9].
 Histonmodifizierungen wirken entweder direkt (cis) durch Beeinflussung der Faltung und Verdichtung der Chromatinfasern oder indirekt (trans) über Rekrutierung von Bindungsproteinen. Mehrere Proteindomänen, die spezifisch unterschiedliche Histonmodifizierungen erkennen, konnten bisher identifiziert werden. (B und C) Bindungsproteine methylierter Lysinreste in H3 und H4, die von der Arbeitsgruppe am Göttinger Max-Planck-Institut beschrieben wurden. Diese haben entweder sogenannte Chromo-Domänen (Heterochromatinprotein 1, HP1; Polycomb, Pc) oder malignant brain tumor (MBT)-Domänen (z. B. L3MBTL1). Die erkannten modifizierten Aminosäuren sowie deren Sequenzumgebung sind schematisch gezeigt. Außerdem sind die dreidimensionalen Strukturen der HP1-, Pc- und L3MBTL1-Proteine, die von den Göttinger Wissenschaftlern in Zusammenarbeit mit anderen Gruppen bestimmt werden konnten, schematisch dargestellt.")
Die Beschreibung der Zusammenhänge zwischen bestimmten Histonmodifizierungen und definierten Bindungsproteinen stellt nur einen ersten Schritt im Verständnis der molekularen Wirkmechanismen der Histonmodifizierungen und ihrer Rolle bei der Entstehung von Chromatindomänen dar. Zur Untersuchung der übergeordneten Arbeitsweise der Bindungsproteine haben die Forscher am Göttinger Max-Planck-Institut nun begonnen, Chromatindomänen im Reagenzglas nachzubilden. In der Tat konnte gezeigt werden, dass HP1 in Abhängigkeit der H3K9me-Modifizierung die Aggregierung und Kompaktierung eines kurzen (sog. oligonukleosmalen) Chromatinabschnitts steuern kann (Abb. 4).
Ausblick
 HP1 führt in Abhängigkeit der von diesem Protein erkannten Histonmodifizierung (Histon Lysin 9-Trimethylierung, H3K9me3) eine Aggregierung rekombinanter Chromatinfasern herbei. Diese Funktionalität ist von der Dimerisierung des HP1-Proteins abhängig, da eine mutante Form (HP1 I161A), die nicht mehr dimerisieren kann, Chromatin nicht aggregieren kann. (B) HP1 führt zu einer Verdichtung rekombinanter Chromatinfasern. Gezeigt sind Aufnahmen individueller, H3K9me3 modifizierter, rekombinanter Oligonukleosome mit dem Atomic-force-Mikroskop mit oder ohne zugegebenes HP1.")

In weiterführenden Arbeiten werden die Forscher sich nun der molekularen Untersuchung von Mustern der Histonmodifizierungen anstelle von individuellen Modifizierungen widmen. Auf der Basis ihrer bisherigen Arbeiten nehmen die Göttinger Wissenschaftler an, dass mehrere Bindungsproteine gleichzeitig mit unterschiedlichen Histonmodifizierungen sowie untereinander wechselwirken, um definierte Chromatindomänen aufzubauen und zu erhalten. Hierzu werden zunehmend komplexere Chromatinsysteme und Untersuchungsmethoden im Reagenzglas entwickelt sowie auf zellulärer Basis analysiert. Von diesen Arbeiten erwarten sich die Forscher nicht nur elementare Erkenntnisse über die Biochemie und molekulare Wirkungsweise der Histonmodifizierungen, sondern erhoffen sich langfristig auch fundamentale Einsichten in die Regulation des Genoms im gesunden sowie kranken Gewebe.