Schwingungsenergietransfer durch molekulare Ketten
Forschungsbericht (importiert) 2003 - Max-Planck-Institut für Multidisziplinäre Naturwissenschaften
Bevor chemische Reaktionen ablaufen können, müssen die Reaktanden in der Regel ausreichend Energie aufnehmen, um Potenzialbarrieren zu überwinden. Bei dieser Aktivierung, die durch Lichtabsorption oder Stöße mit Lösungsmittelmolekülen der Umgebung erfolgen kann, wird die Energie in den Schwingungsfreiheitsgraden der Reaktanden gespeichert. Haben die Reaktanden genügend Energie aufgenommen, erfolgt jedoch die eigentliche Reaktion (zum Beispiel ein Bindungsbruch) im Falle polyatomarer Moleküle nicht augenblicklich, sondern wird durch intramolekulare Energieumverteilungsprozesse verzögert. Die Ursache hierfür besteht darin, dass die Energie über viele Schwingungsfreiheitsgrade des Moleküls verteilt ist und sich erst als Folge einer Fluktuation in der zu brechenden Bindung (der Reaktionskoordinate) konzentrieren muss. Intramolekulare Umverteilung von Schwingungsenergie (intramolecular vibrational energy redistribution, IVR) ist somit ein wichtiger Bestandteil chemischer Reaktionen. Die Annahme von sehr schnellem IVR im Vergleich zum eigentlichen Reaktionsschritt ist von fundamentaler Bedeutung für die Anwendbarkeit statistischer Theorien zur Berechnung von Reaktionsgeschwindigkeitskonstanten. Andererseits eröffnet eine Behinderung der Schwingungsenergieumverteilung durch interne Engpässe Möglichkeiten einer modenselektiven Chemie. Hinter dieser Bezeichnung verbirgt sich zum Beispiel die Hoffnung, mit einem Laser selektiv ausgewählte Bindungen in einem Molekül anzuregen und für eine Reaktion zu präparieren.
 mit einem kurzen Laserpuls auf eine Schwingungstemperatur von etwa 1150 Kelvin angeregt. Die Ausbreitung der Überschussenergie durch die Brücke zur Anthrazenseite (blau) wird mit einem verzögerten Laserpuls durch Messung der Temperatur in beiden Chromophoren zeitlich verfolgt.")
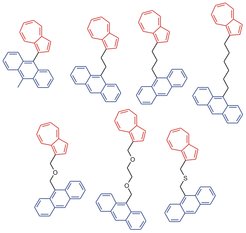
Die Arbeitsgruppe um Dirk Schwarzer beschäftigt uns seit einiger Zeit mit einem speziellen IVR-Prozess, nämlich dem Transfer von Schwingungsenergie durch molekulare Ketten, die an beiden Enden mit relativ großen Farbstoffmolekülen verbunden sind. Die beiden Farbstoffmoleküle repräsentieren Wärmebäder mit zunächst unterschiedlichen Temperaturen, die sich mit der Zeit angleichen. Somit ähnelt ein solcher Versuch einem klassischen Wärmeleitungsexperiment, bei dem zwei Wärmereservoirs unterschiedlicher Temperaturen über einen Stab verbunden sind. Ein Ziel dieser Arbeiten ist es herauszufinden, ob und unter welchen Bedingungen Fouriers Gesetz der Wärmeleitung auf molekularer Ebene anwendbar ist. Im Gegensatz zur Wärmeleitung in einer makroskopischen Probe verläuft die Schwingungsenergieumverteilung im Molekül auf der extrem kurzen Zeitskala von Pikosekunden (1 ps = 10-12 s) oder darunter, was den Einsatz der Ultrakurzzeit-Spektroskopie erforderlich macht.
Für die Untersuchungen verwenden die Göttinger Wissenschaftler die in Abbildung 1 dargestellten verbrückten Azulen-Anthrazen-Verbindungen. Im Experiment wird der Azulenteil des Moleküls mit einem kurzen
Laserpuls selektiv angeregt. Die Schwingungsanregung entspricht einer Aufheizung des Azulen-Chromophors auf 1150 Kelvin (K). Anschließend wird die Ausbreitung der Überschussenergie durch die Brücke auf das gesamte Molekül vermessen, indem man die Energien des Azulen- und/oder des Anthrazenteils mit einem verzögerten Laserpuls abtastet. Die Messmethode beruht auf der Tatsache, dass sich elektronische Absorptionsspektren mit zunehmender Schwingungsenergie verbreitern und zu niedrigeren Energien hin ausdehnen. Wird die Wellenlänge des Abtastlaserpulses auf die langwellige Flanke eines solchen elektronischen Übergangs abgestimmt, so äußert sich die Zunahme der Schwingungsenergie in einer verstärkten Absorption des betreffenden Laserlichtes. Im Falle von Azulen und Anthrazen sind die energieabhängigen Absorptionsspektren bekannt, sodass man die gewonnenen Absorptions-Zeit-Signale direkt in Energie-Zeit-Kurven übersetzen kann.
, Anthrazen (blau) und der verbrückten Verbindung Azulen-(CH2)6-Anthrazen (schwarz). Durch geeignete Wahl der Wellenlänge des Abtastlaserpulses kann selektiv die Azulen- (roter Pfeil) bzw. Anthrazenenergie (blauer Pfeil) spektroskopisch vermessen werden.")
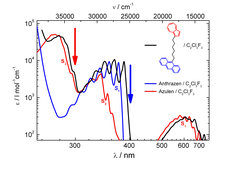
Abbildung 2 zeigt beispielhaft für die Verbindung Azulen-(CH2)6-Anthrazen, dass die Absorptionsbanden beider Chromophore auch in den kombinierten Molekülen wohlsepariert sind. So ist es möglich, selektiv die Energie des Azulenchromophors auf der langwelligen Flanke der S3-Absorptionsbande bei etwa 300 nm abzufragen (roter Pfeil) oder unter Einsatz einer Wellenlänge von 400 nm den Energieinhalt des Anthrazenteils im Bereich der S1-Bande (blauer Pfeil) zu vermessen.
; intramolekularer Energiefluss und anschließende Stoßdesaktivierung beim Azulen-(CH2)3-Anthrazen (Abtasten der Azulen- (Mitte) bzw. Anthrazenenergie, unten).")
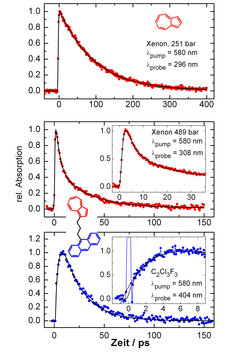
In Abbildung 3 sind experimentell beobachtete Absorptions-Zeit-Profile dargestellt. Das obere Signal zeigt zunächst eine Kontrollmessung des unverbrückten Azulenmoleküls gelöst in flüssigem Xenon. Mit der Laseranregung zur Zeit t = 0 beobachtet man einen sprunghaften Anstieg der Absorption, der auf die Zunahme der Schwingungsenergie zurückzuführen ist. Infolge von Stößen mit den umgebenden Xenonatomen wird diese Energie aber an die Umgebung abgegeben. Dieser Prozess des stoßinduzierten intermolekularen Schwingungsenergietransfers verläuft annähernd exponentiell mit einer Zeitkonstanten von 95 ps. Im Gegensatz dazu erfolgt der Energieabfall im Azulenteil der verbrückten Verbindungen qualitativ verschieden. Das ist am Beispiel der Verbindung Azulen-(CH2)3-Anthrazen in Abbildung 3 dargestellt (siehe mittleres Signal). Anstelle eines einfach exponentiellen Abfalls beobachtet man, dass die Energie auf zwei separaten Zeitskalen aus dem Azulen abfließt. Die schnellere Komponente mit einer Zeitkonstanten von τIVR = (3.7±0.3) ps ist auf den intramolekularen Schwingungsenergietransfer durch die Kette zum Anthrazenteil des Moleküls zurückzuführen. Nach 15 ps ist der IVR-Prozess abgeschlossen, die Schwingungsenergie ist innerhalb des Moleküls vollständig äquilibriert und wird nun auf einer Zeitskala von etwa 40 ps an das umgebende Medium abgegeben. Diese Interpretation wird unterstützt durch die Tatsache, dass dieselben intra- und intermolekularen Energietransfer-Zeitskalen auch auf der Anthrazenseite zu beobachten sind. Wie im unteren Teil von Abbildung 3 gezeigt ist, steigt die Schwingungsenergie im Anthrazenteil nach Anregung der Azulenseite mit einer Zeitkonstanten von ~4 ps an, gefolgt von einem Abfall, der annähernd 100 ps dauert.
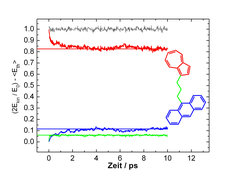
Eine genauere Analyse des Messsignals um den Zeitnullpunkt ergibt, dass dem 4 ps-Anstieg eine wesentlich schnellere Komponente vorgelagert ist, die unter der Zeitauflösung des Experimentes von etwa 0.3 ps liegt. Dieser extrem schnelle Beitrag ist folgendermaßen zu erklären: Die anfängliche lokale Schwingungsanregung des Azulens kann näherungsweise als Überlagerung von harmonischen Normalschwingungen des Gesamtmoleküls betrachtet werden. Da die Normalschwingungen mit unterschiedlichen Frequenzen oszillieren, geraten sie nach kurzer Zeit außer Phase, sodass sich die Schwingungsamplitude am Azulen verringert und auf das gesamte Molekül ausbreitet. Dieser Prozess ist nicht mit Umverteilung von Schwingungsenergie innerhalb der Normalmoden des Gesamtmoleküls verbunden und stellt deshalb eine harmonische Komponente zur Energieäquilibrierung dar. Mithilfe quantenchemischer Rechnungen lassen sich die Normalschwingungen des Moleküls bestimmen, mit denen anschließend der harmonische Energiefluss simuliert werden kann. Das Ergebnis ist in Abbildung 4 dargestellt. Man erkennt, dass innerhalb von 0.3 ps etwa 18% der ursprünglichen Anregungsenergie aufgrund dieses Mechanismus aus dem Azulenchromophor abfließt; 12% erreichen den Anthrazenteil. Erst danach beginnt der eigentliche IVR-Prozess, der zum internen Energiegleichgewicht führt, gefolgt von Stoßdesaktivierung und Thermalisierung mit dem umgebenden Medium.
3-Anthrazen. Aufgetragen sind Änderungen der kinetischen Energie während der zeitlichen Entwicklung der Normalschwingungen nach lokaler Anregung des Azulens (rot: Energie im Azulenteil; blau: im Anthrazenteil; grün: in der Kette; schwarz: Summe aller Beiträge).")
In Abbildung 5 sind die gemessenen IVR-Zeiten τIVR gegen die Länge der Ketten (ausgedrückt durch die Anzahl chemischer Bindungen n) aufgetragen, welche die Farbstoffe verbindet. Für kurze Brücken findet man einen linearen Anstieg von τIVR mit der Kettenlänge. Dieses Verhalten könnte man konsistent mit dem Fourierschen Wärmeleitungs-Gesetz beschreiben, wonach der Energiefluss durch den Wärmeleitkoeffizienten κ des Leiters charakterisiert wird und entgegen dem Temperaturgradienten über der Kette erfolgt. Da in unserem Experiment der Temperaturgradient mit zunehmender Kettenlänge abnimmt, vergrößert sich die Zeitspanne, die die interne Energieäquilibrierung benötigt. Für n ≥ 4 beobachtet man allerdings (siehe Abb. 5), dass die IVR-Zeiten annähernd konstant sind. Dabei sind sie für aliphatische Brücken etwas kürzer als für (Thio)ether. Wollte man dieses Verhalten mit dem Wärmeleitungsgesetz beschreiben, müsste der Wärmeleitkoeffizient der Kette proportional zu deren Länge ansteigen. Das lässt bereits vermuten, dass Fourier's Gesetz der Wärmeleitung in diesem Fall nicht anwendbar ist.
 von der Länge der Brücke, die die Chromophore verbindet (rot: aliphatische Ketten; blau: Ether; grün: Thioether).")

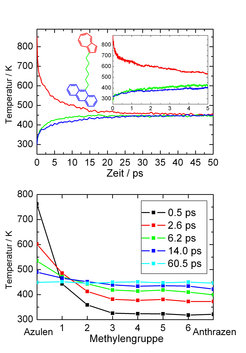
Um den Mechanismus des intramolekularen Energieflusses im Detail zu verstehen, führten Dirk Schwarzer und sein Team molekulardynamische Computersimulationen durch. Dazu beschreibt man die innermolekularen Kräfte durch ein vieldimensionales Kraftfeld, in dem die Bewegungen der Teilchen durch Lösen der klassischen Bewegungsgleichungen bestimmt werden. In Übereinstimmung mit dem Experiment findet man, dass der innermolekulare Energiefluss in zwei Schritten erfolgt (siehe Abb. 6, oben). Nach der Anregung verliert der Azulenteil innerhalb von 0.3 ps 20% der Anregungsenergie aufgrund des oben beschriebenen harmonischen Energieflusses. Danach beginnt der IVR-Prozess, der zur Energieäquilibrierung innerhalb des Moleküls führt. Die zugehörigen Zeitkonstanten sind zwar um etwa 50% größer als die experimentellen (was auf fehlerhafte Potenziale und die Vernachlässigung der Quantennatur der Schwingungsbewegung zurückzuführen ist), aber das in Abbildung 5 beobachtete Sättigungsverhalten von τIVR für Ketten mit n ≥ 4 wird qualitativ reproduziert. Dies lässt vermuten, dass in Experiment und Computersimulation die gleichen Mechanismen für den Energieaustausch zwischen den Farbstoffen sorgen.
Die molekulardynamische Simulation erlaubt es, diese Mechanismen aufzuklären. In Abbildung 6 sind unten während der Relaxation auftretende Temperaturprofile von der Azulenseite (links) durch die Kette zur Anthrazenseite (rechts) aufgetragen. Wie zu erwarten gibt es nach der Anregung einen großen Temperaturgradienten zwischen dem Azulenteil und der Brücke. Dieser Gradient wird mit der Zeit zwar kleiner, aber erstaunlicherweise bleibt er an der Verbindung des Azulenchromophors zur Kette lokalisiert, während er innerhalb der Kette verschwindet. Für Moleküle mit kürzeren Ketten findet man auch große Temperaturgradienten an der Anthrazenseite. Offenbar wird der Energiefluss durch die Ankopplung der Schwingungsbewegung der Farbstoffe an die Brücke limitiert. In der Kette erfolgt der Energietransport wesentlich effizienter, sodass die Zeit für die intramolekulare Energieäquilibrierung ab n ≥ 4 unabhängig von der Kettenlänge wird. Die Rechnungen zeigen, dass der Energiefluss nicht wie im Fall klassischer Wärmeleitung diffusiv erfolgt, sondern ohne Streuung der für den Transport relevanten Normalschwingungen.
6-Anthrazen (oben: Temperaturen in verschiedenen Teilen des Moleküls als Funktion der Zeit; unten: Temperaturprofile in der Brücke einschließlich dem Azulen- (links) und Anthrazenchromophor (rechts).")
Literatur
Schwarzer, D., C. Hanisch, P. Kutne and J. Troe: "Vibrational energy transfer in highly excited bridged azulene-aryl compounds: direct observation of energy flow through aliphatic chains and into the solvent. Journal of Physical Chemistry A 106, 8019-8028 (2002).
Schwarzer, D., P. Kutne, C. Schröder and J. Troe: "Intramolecular vibrational energy redistribution in bridged azulene-anthracene compounds: ballistic energy transport through molecular chains. Journal of Chemical Physics 121, 1754-1764 (2004).