Neue Ansätze zu Modellierung und Behandlung der neurodegenerativen Prozesse der Schizophrenie
Forschungsbericht (importiert) 2008 - Max-Planck-Institut für Multidisziplinäre Naturwissenschaften
Einleitung
Die Division "Klinische Neurowissenschaften" beschäftigt sich seit über 15 Jahren mit der Entwicklung neuer Therapiekonzepte bei bislang unheilbaren neurologischen und psychiatrischen Krankheiten. Mit diesem Forschungsschwerpunkt stellt sie den translationalen, d.h. an der Übersetzung zum Menschen arbeitenden, klinikorientierten Teil des neurowissenschaftlich ausgerichteten Max-Planck-Instituts für experimentelle Medizin dar. Bei den entwickelten Therapiekonzepten handelt es sich um neue psychotherapeutische ebenso wie innovative pharmakologische Verfahren. Als besonders erfolgreich erwies sich in den letzten zehn Jahren ihre Forschungsarbeit mit Erythropoietin (EPO).
EPO ist ein hämatopoietischer Wachstumsfaktor, von welchem man bereits seit Jahrzehnten weiß, dass er in Niere und fetaler Leber exprimiert wird. Seine Bildung im Gehirn und seine vielschichtige Rolle während der Hirnentwicklung jedoch sind erst im letzten Jahrzehnt zunehmend erkannt worden [1; 2]. Die Expression von EPO wird hauptsächlich über hypoxia inducible factor 1 (HIF-1) gesteuert, es gibt aber auch Hinweise auf andere Induktionsmechanismen. EPO ist ideal geeignet für translationale Ansätze, da es sich seit über 20 Jahren zur Behandlung von Patienten mit Blutarmut unterschiedlicher Genese im klinischen Gebrauch befindet. Dabei erwies sich EPO stets als sehr gut verträglich und sicher. Insbesondere die in zahlreichen Publikationen unterschiedlicher Arbeitsgruppen gezeigten Eigenschaften am Nervensystem machen EPO zu einem vielversprechenden Kandidaten für den Einsatz im Rahmen innovativer neuroprotektiver/neuroregenerativer Behandlungsstrategien. EPO wirkt anti-apoptotisch, anti-oxidativ, anti-entzündlich, neurotroph und plastizitätsmodulierend, zusätzlich auch stimulierend auf Neurogenese und Differenzierung von Nervenzellen. EPO wurde in mittlerweile weit über 100 präklinischen Publikationen als wirksame neuroprotektive Substanz beschrieben. Dabei kamen Tiermodelle ganz unterschiedlicher neuropsychiatrischer Erkrankungen, von Ischämie/Hypoxie über Schädel-Hirn-Verletzung und Spinaltrauma bis hin zu neurodegenerativen und entzündlichen Erkrankungen, zum Einsatz, in welchen fast ausnahmslos von einer Verbesserung klinischer wie auch biochemischer oder histologischer Parameter berichtet wird [1; 3].
Lange vor Erscheinen der genannten Publikationen, nämlich bereits im Jahre 1997, begannen die Göttinger Forscher, basierend auf eigenen präklinischen Daten, EPO als neuroprotektive Behandlungsstrategie bei Patienten mit ischämischem Schlaganfall einzusetzen: mit Erfolg [4]. Es folgten erste Therapiestudien bei so unterschiedlichen Krankheiten wie Schizophrenie [5], Multiple Sklerose [6] oder Demenz/Depression. Bis heute sind die Wissenschaftler um Hannelore Ehrenreich in diesem Feld einsame Pioniere. Zwar laufen inzwischen, aufbauend auf diesen Arbeiten, weltweit etwa 40 klinische EPO-Studien mit neuroprotektiver Indikation, Wirksamkeitsdaten sind jedoch noch nicht publiziert [1]. Wie an den ätiologisch/pathogenetisch sehr unterschiedlichen Krankheiten erkennbar, ist es nicht das Ziel, krankheitsspezifisch vorzugehen, sondern vielmehr Determinanten der so genannten gemeinsamen Endstrecke zu modulieren, welche vielen dieser Krankheiten zugrunde liegen. Diese Determinanten der gemeinsamen Endstrecke führen zum Fortschreiten der Erkrankung, zur Verschlechterung der Symptomatik und zu progredienten Funktionsausfällen. Beispiele dafür sind neuronale Plastizitätsveränderungen bis hin zu vermehrter Nervenzell-Apoptose, Entzündung, oxidativer Stress, metabolische Dysfunktion oder vaskuläre Insuffizienz. Wie bereits geschildert hat EPO Eigenschaften, die es im Hinblick auf viele dieser Endstreckenmechanismen als sehr wirksam erscheinen lassen.
Im Nervensystem wirkt EPO über spezifische Rezeptoren, EPOR, welche während der intrauterinen Entwicklung um ein Vielfaches höher exprimiert werden als im adulten Hirn. Postnatal kommt es schrittweise zu einer Reduktion der Expression bis hin zu niedrigen Werten beim gesunden Erwachsenen [7]. Diese Situation ändert sich jedoch rasch, wenn schädigende Einflüsse auf das Nervensystem zu einer „Notlage“ von Nervenzellen führen, wie beispielsweise Hypoxie, Ischämie, Entzündungsvorgänge unterschiedlichster Genese oder degenerative Prozesse. So wurde etwa nach Ischämie/Hypoxie, aber auch bei neurodegenerativen Erkrankungen wie Morbus-Alzheimer eine erhöhte EPOR- Expression in verschiedenen Zellpopulationen (Neurone, Glia, Endothelzellen) gefunden. Wird andererseits im Schlaganfall-Tierexperiment durch intracerebroventrikuläre Gabe von löslichem EPOR endogenes EPO massiv weggefangen, so kommt es zu einer dramatischen Schadensvermehrung. Diese Beobachtungen ließen EPO/EPOR als ein wichtiges endogenes protektives „Stand-by“-System im adulten Gehirn erkennen.
Schizophrenie als neurodegenerative Erkrankung
Viele Jahre stritten sich Schulen in der Psychiatrie über eine mögliche Beteiligung neurodegenerativer Prozesse an der Pathophysiologie der Schizophrenie. Insbesondere die Abwesenheit von Astrogliose (Wuchern von Astrocyten), einem obligatorischen Anzeichen für Neurodegeneration, in den Gehirnen verstorbener schizophrener Patienten ließ Neuropathologen noch vor wenigen Jahrzehnten zu dem festen Schluss kommen, dass degenerative Prozesse bei Schizophrenie ausgeschlossen seien. Einig war man sich darüber, dass Entwicklungsdefekte an der Entstehung dieser Erkrankung beteiligt sind, nachgewiesen durch veränderte Größe von Nervenzellen, Migrationsstörungen von Neuronen in der Großhirnrinde oder andere cytoarchitektonische Anomalien. Mit der ausschließlichen Interpretation der Schizophrenie als Entwicklungsstörung war automatisch deren Bedeutung als Hirnerkrankung in Frage gestellt. So erklärt sich auch die Tatsache, dass die neurobiologische Grundlagenforschung im Bereich der Schizophrenie im Gegensatz zu anderen neurodegenerativen Erkrankungen noch in den Kinderschuhen steckt. Verwunderlich ist diese Entwicklung deshalb, weil bereits vor über 100 Jahren, aufbauend auf der französischen Literatur des frühen 19. Jahrhunderts, Emil Kraepelin, der Vater der modernen Psychiatrie, den Begriff „Dementia praecox“ in sein Klassifikationssystem aufgenommen hat. Allein der Begriff Demenz beinhaltet das Vorliegen eines degenerativen Prozesses, da mit ihm der Verlust einmal erworbener kognitiver Funktionen ausgedrückt wird. Heutzutage hat sich auf der Basis der modernen Bildgebung die Erkenntnis um die neurodegenerativen Aspekte der Schizophrenie deutlich verbessert. So wurde von Thompson und Mitarbeitern im Jahre 2001 eine prospektive 5-Jahresstudie an Patienten mit juvenilem Beginn der Schizophrenie publiziert [8]. Diese Studie zeigte erstmals auf der Basis moderner voxelbasierter morphometrischer Analyse des Gehirns, dass es zu Beginn der Erkrankung zu einem progredienten Verlust an grauer Substanz im Bereich des Parietallappens kommt, der sich sukzessive nach temporal und frontal ausbreitet. Damit war proof-of-principle gezeigt, dass bei der Schizophrenie, möglicherweise angestoßen durch gestörte Entwicklungsvorgänge, auch neurodegenerative Prozesse ablaufen.
Entwicklung eines Tiermodells der Neurodegeneration bei Schizophrenie
Dieser Befund veranlasste die Wissenschaftler am Göttinger Institut, ein Tiermodell zu entwickeln, in welchem die neurodegenerativen Aspekte der Schizophrenie modelliert werden sollten. Bei dem Design des Modells gingen sie von zwei wesentlichen Annahmen aus: (1) Der Parietallappen ist der entscheidende Ausgangspunkt für die geschilderten neurodegenerativen Prozesse und (2) Juveniles Alter ist Voraussetzung für den Prozessbeginn. Beide Annahmen erwiesen sich als grundlegend. Obwohl bei der Schizophrenie die endogene Ursache für den Startpunkt der Degeneration in der Pubertät und im Parietallappen noch völlig unklar ist, ahmen die Forscher mit ihrem Modell den Beginn des degenerativen Prozesses exogen nach, indem sie bei juvenilen (28 Tage alten) Mäusen stereotaktisch durch die intakte Schädeldecke hindurch eine Kryoläsion (Applikation von -80° Kälte über 60 Sekunden mittels Metallspitze) im rechten Parietallappen setzen [9]. In der Tat wird dadurch eine progrediente bilaterale Hirnatrophie ausgelöst, begleitet von Verhaltensänderungen sowie Lern- und Gedächtnisstörungen, wie sie ähnlich bei der Schizophrenie bekannt sind (Abb. 1 und 2).
-Bilder. Beachtenswert sind die allseits erweiterten Ventrikel nach Läsion und die normale Hirnkonfiguration (vergleichbar den scheinoperierten Tieren) bei lädierten und EPO-behandelten Mäusen.")
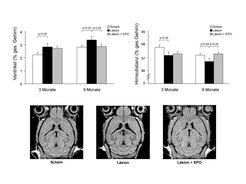
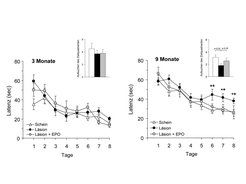
Interessanterweise kommt es mit zunehmendem Alter der Tiere zu einer deutlichen Zunahme der kognitiven Defizite, sodass die Wissenschaftler davon ausgehen, dass zusätzlich zur gesetzten Vorschädigung der normale Alterungsprozess eine additive Rolle im Sinne einer Dekompensation spielt (Abb. 2).
, bei scheinoperierten, lädierten und lädierten + EPO-behandelten Tieren, 3 und 9 Monate nach parietaler Kryoläsion. Während sich zum Zeitpunkt \"3 Monate\" keine Unterschiede zeigen, wird nach 9 Monaten die deutlich schlechtere Lernleistung lädierter Tiere sichtbar. EPO-Behandlung kann den Läsionseffekt komplett verhindern.")
Da die molekularen und zellulären Grundlagen der globalen Hirnatrophie unklar waren, führten die Wissenschaftler ein umfassendes Projekt durch, in welchem sie auf stereologischer Basis verschiedene Zelltypen im Gehirn der kryolädierten Mäuse quantifizierten [10]. Sie fanden mit allen angewandten Methoden bei allen untersuchten Parametern von Bildgebung über Histologie bis zum Western Blot, dass die unilateral gesetzte Läsion stets bilaterale Veränderungen im Gehirn induziert. Als erstes prominentes Ereignis zeigte sich schon nach wenigen Stunden eine bilaterale Zunahme der Mikroglia, welche auch noch zwölf Monate nach der Läsion nachweisbar blieb, passend zu einem chronisch persistierenden, milden inflammatorischen Prozess. Während die Gesamtzahl von Neuronen und Astrozyten in Gyrus cinguli und Hippocampus unverändert war, also auf das Vorliegen einer nicht-gliotischen Neurodegeneration hinweist (wie sie bei der Schizophrenie zu vermuten ist), fanden die Forscher quantitative Verschiebungen von Nervenzellsubpopulationen. Parvalbumin-positive inhibitorische GABAerge Interneurone waren bilateral im Hippocampus ebenso vermehrt wie die Expression des GABA-synthetisierenden Enzyms GAD67. Im Gefolge der parietalen Läsion war weiterhin eine Reduktion des präsynaptischen Proteins Synapsin1 feststellbar, vereinbar mit einer Beeinträchtigung von Synapsenfunktion und Neuroplastizität. Eine verminderte Expression des Myelinproteins CNP-ase (cyclic nucleotide phosphodiesterase) spiegelt nicht nur eine Abnahme der Oligodendrozytenzahl wider, sondern dürfte auch zu der beobachteten Hirnatrophie beitragen.
Besonders bemerkenswert ist, dass eine frühe, d.h. unmittelbar nach Setzen der Läsion beginnende, sich über zwei Wochen erstreckende Behandlung mit hochdosiertem EPO alle geschilderten Veränderungen vollständig zu verhindern vermag, sowohl Atrophie und Verhaltensänderungen als auch morphologisch-histologische Folgeerscheinungen der parietalen Läsion. Aus diesen Befunden lassen sich drei Schlüsse ziehen: (1) Eine unilaterale Parietallappenläsion bei juvenilen Mäusen induziert eine bilaterale, nicht-gliotische Neurodegeneration, welche einer frühen EPO-Behandlung zugänglich ist. (2) Die hier beobachtete nicht-gliotische Neurodegeneration scheint wesentliche Merkmale der Neurodegeneration bei der Schizophrenie nachzustellen. (3) Für die Initiation dieses neurodegenerativen Prozesses ist ein Setzen der Läsion zu einer vulnerablen Zeit (Pubertät) und in einer vulnerablen Region (Parietallappen) notwendig. Läsionen anderen Orts (z.B. okzipital) oder in späterem Alter (z.B. 3 Monate, 12 Monate) zeigen keine derartigen globalen Folgen am Gehirn [9; 10].
Erste neuroprotektive Therapie-Ansätze bei der Schizophrenie
Neuroprotektion/Neuroregeneration bei Schizophrenie liegt dann nahe, wenn neurodegenerative Prozesse bei dieser Krankheit angenommen werden, die abgeschwächt oder verhindert werden sollen. Ein solcher Ansatz ist bei dieser Erkrankung völlig neu. Stimuliert durch die potenten Effekte von EPO im beschriebenen Tiermodell – im Hinblick auf Verhinderung von Hirnatrophie, Verhaltensauffälligkeiten und nicht-gliotischer Neurodegeneration – bereiteten die Göttinger Wissenschaftler schon 2001 eine neuroprotektive Studie mit EPO bei schizophrenen Patienten vor [11]. Wichtigste Frage war dabei zunächst, ob EPO, als ein großes Molekül von über 30 000 Dalton, in der Lage sein würde, eine bei Schizophrenen zumeist intakte Blut-Hirn-Schranke zu penetrieren. Diese Frage beantworteten sie, indem sie Indium111-markiertes EPO intravenös jungen, männlichen, schizophrenen Patienten und gesunden Kontrollprobanden verabreichten. die dann mittels SPECT (single photon emission computed tomography) auf die Verteilung von EPO im Organismus und insbesondere im Gehirn hin untersucht wurden. Es zeigte sich, dass schizophrene Patienten eine deutliche Anreicherung von Indium111-Signalen im Hirn aufwiesen, wobei allerdings auch gesunde Kontrollen erkennen ließen, dass eine Penetration der intakten Blut-Hirn-Schranke für EPO möglich ist (Abb. 3) [11].
 intracerebral bei Schizophrenen im Vergleich zu Gesunden. Dies bildet sich analog in den signifikanten Unterschieden der Mittelwerte ab.")
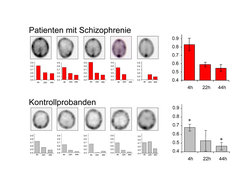
Wichtig ist es zu betonen, dass der Prozentsatz von EPO, welcher ins Gehirn gelangt, nur bei etwa 0.1-1% des peripher verabreichten EPO liegt. Deshalb erfordern neuroprotektive Indikationen hohe EPO-Dosen. Wie konnte nun die Mehranreicherung im Hirn schizophrener Probanden erklärt werden? Die Antwort zu dieser Frage ergab sich durch Post-mortem-Untersuchungen an den Gehirnen Schizophrener versus Gesunder: In Hippocampus und Cortex schizophrener Menschen war eine signifikant höhere EPOR-Expression sowohl durch Neurone als auch durch Gliazellen nachweisbar. Dadurch war die stärkere Anreicherung des Indium111-markierten EPO in den Gehirnen dieser Patienten erklärbar. Warum es jedoch zu einer Erhöhung der Rezeptorexpression kommt, ist augenblicklich unklar. Höchstwahrscheinlich lässt sie sich aber auf die bereits geschilderte „metabolische Notlage“ entsprechender Hirnzellen zurückzuführen, welche mit einer Steigerung der EPOR-Expression beantwortet wird und auf die Präsenz des endogenen protektiven EPO-Systems (vielleicht mit relativem Defizit von EPO) hinweist. Dennoch stellte sich die Frage, ob möglicherweise Antipsychotika (als zusätzlich zur Krankheit selbst vorhandenes Unterscheidungsmerkmal zwischen Patienten und Kontrollen) Einfluss auf die EPOR-Expression nehmen könnten. Während die Wissenschaftler darauf keine eindeutige Antwort fanden, konnten sie aber in ausgedehnten Untersuchungen an primären hippocampalen Neuronenkulturen zeigen, dass ein durch Haloperidol induzierter Zelltod mit EPO signifikant reduziert wird [11].
Somit war der Boden bereitet für die Durchführung der ersten multizentrischen, doppelblinden, randomisierten, placebokontrollierten Proof-of-Principle-Studie (Phase IIb) mit EPO als neuroprotektiver/neuroregenerativer Add-on-Therapie bei schizophrenen Patienten [5]. Obwohl Patienten mit der ersten Episode einer schizophrenen Psychose, d. h. einem insgesamt sehr viel aggressiveren Krankheitsgeschehen, mit zu diesem Zeitpunkt besonders dramatischer, progredienter, kognitiver Verschlechterung und zunehmender Hirnatrophie, für den neuroprotektiven Therapieansatz sicherlich die viel besser geeignete Zielpopulation gewesen wären, nahmen die Göttinger Forscher das Risiko auf sich, zunächst chronisch kranke, stabil kognitiv beeinträchtigte Patienten einzuschließen. Sie taten dies vornehmlich aus ethischen Überlegungen heraus (Langzeit-Compliance, Einwilligungsfähigkeit, legale Situation bezüglich möglicher Betreuung, erste Durchführung einer Proof-of-Principle-Studie bei dieser Zielgruppe). Zusätzlich zu diesen ethischen Überlegungen kam jedoch auch die Hypothese, dass prinzipiell jedes Hirn eine messbare Fähigkeit zur Regeneration haben müsste.
Die Gesamtdauer der ersten Proof-of-Principle-EPO-Schizophrenie-Studie erstreckte sich über zwei Jahre mit einer individuellen Dauer von zwölf Wochen nach Studienbeginn. Chronisch schizophrene Männer mit definiertem kognitiven Defizit, gleichbleibender Medikation und stabilem Krankheitszustand, wurden drei Monate lang add-on mit wöchentlichen Kurzinfusionen von je 40 000 I.E. EPO oder Placebo behandelt. Wichtigster Ergebnisparameter waren schizophrenierelevante kognitive Funktionen zum Zeitpunkt der Therapiebeendigung (Woche 12). Ein neuropsychologischer Summenscore (Repeatable Battery for the Assessment of Neuropsychological Status, mit den Subtests Delayed Memory, Language Semantic Fluency und Attention, sowie dem Wisconsin Card Sorting Test) wurde vor Behandlungsbeginn (baseline) und danach unter EPO/Placebo-Behandlung in den Wochen 2, 4 und 12 der Studienteilnahme ermittelt. Sowohl Placebo- als auch EPO-Patienten verbesserten sich in allen evaluierten Kategorien, Neuropsychologie, Psychopathologie und Sozialfunktionen. Patienten, welche EPO erhielten, zeigten jedoch ein signifikant besseres Ergebnis als Patienten unter Placebo im Hinblick auf ihre kognitive Leistungsfähigkeit (Abb. 4).

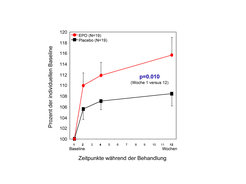
Auswirkungen der EPO-Behandlung auf Psychopathologie oder soziale Funktionsparameter waren nach dieser kurzen Behandlungszeit (bei chronisch, d. h. schon über zehn Jahre kranken Patienten) nicht feststellbar. Interessanterweise kam es unter EPO-Gabe zu einem signifikanten Abfall der Serumspiegel von S100B, einem glialen Schadensmarker [5]. In einer kürzlich abgeschlossenen, umfangreichen, voxelbasierten morphometrischen Analyse, in welcher Hirndimensionen von EPO- versus Placebo-Patienten vor und nach der Behandlung verglichen werden, zeigte sich überraschenderweise bereits nach nur zwölf Wochen Behandlung, dass die EPO-Gruppe, verglichen mit der Kontrollgruppe, einen geringeren Abbau an grauer Substanz in schizophrenierelevanten Hirnregionen aufweist. Da der Verlust an grauer Substanz pro Jahr bei Schizophrenen etwa 0,5% (im Vergleich zu Gesunden mit etwa 0,2%) beträgt, wird die Messbarkeit einer reduzierten Hirnsubstanzminderung unter neuroprotektiver Behandlung mit dieser sehr subtilen Meßmethode plausibel.
Damit konnten die Göttinger zeigen, dass EPO die erste Substanz ist, mit welcher es gelingt, selbst bei chronisch kranken Schizophrenen den progredienten Verlust an grauer Substanz zu reduzieren und gleichzeitig kognitive Funktionen zu verbessern. Diese Befunde bedeuten nicht den Abschluss, sondern erst den Anfang einer neuen Entwicklung im Bereich der Schizophrenietherapie-Forschung. Nachdem eine Einflussnahme auf Kognition und Hirnatrophie bislang therapeutisch praktisch undenkbar war, sollte auf der Basis dieser Proof-of-Concept-Studie mit Hochdruck an der Verbesserung des Konzepts gearbeitet werden. Dazu gehören etwa die Austestung unterschiedlicher EPO-Dosen, aber auch von EPO-Varianten, Modulation der Behandlungsfrequenz und -dauer, Kombination mit verschiedenen antipsychotischen Substanzen, sicherlich aber auch die Übersetzung dieser Befunde auf die eigentliche Zielpopulation, nämlich Patienten mit erster Episode einer schizophrenen Psychose. Zu diesem frühen Zeitpunkt der Erkrankung laufen die stärksten Veränderungen im Hinblick auf Kognition und graue Substanz ab. Demnach erscheint es sinnvoll, genau jene Zielgruppe mit hoher Priorität zu untersuchen. Sollte es gelingen, kognitiven Leistungsabfall und Verlust an grauer Substanz bei jungen, ersterkrankten Patienten zu reduzieren, so hätte dies nicht nur individuelle, sondern auch ganz erhebliche gesellschaftliche Auswirkungen.
Zurück zur Maus: Erste Untersuchungen zu EPO-Wirkmechanismen im Hinblick auf kognitive Funktionen
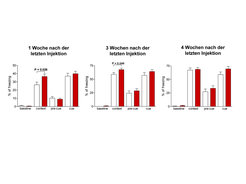
Nach den ermutigenden Ergebnissen sogar bei chronisch schizophrenen Patienten planen Hannelore Ehrenreich und ihr Team nicht nur die Fortführung einer klinischen Proof-of-Principle-Studie bei Patienten mit erster Episode einer Schizophrenie mit dem Ziel, kognitiven Leistungsabfall und Verlust an grauer Substanz einzudämmen, sondern sind gleichzeitig zurückgegangen zur Maus, um zu untersuchen, welche Mechanismen die Effekte auf die Kognition erklären könnten [12]. Sie führten dazu eine große Zahl unterschiedlicher Experimente mit Verhaltenstests bei Mäusen durch, in welchen höhere Hirnfunktionen untersucht werden. Bei diesem ersten mechanistischen Ansatz fokussierten sie auf gesunde, junge Mäuse, um potenzielle Interferenzen mit Krankheitsvariablen zunächst auszuschalten. Nach einer Reihe positiver Befunde mit EPO-Behandlung bei teilweise sehr komplexen kognitiven Aufgaben, etwa Verbesserung der Leistungsfähigkeit im Five Choice Serial Reaction Time Task oder im Morris Water Maze Test, entwickelten sie für die weiteren Versuche ein Modell, in welchem es zu einer robusten, hoch reproduzierbaren und rasch evaluierbaren kognitiven Verbesserung durch EPO kommt. Sie bedienten sich des so genannten Fear Conditioning, in welchem die Tiere auf Context und Cue, d. h. eine definierte Umgebung und einen akustischen Schlüsselreiz aversiv durch Elektroschock konditioniert werden. Werden die Mäuse im Gefolge dieser Konditionierung entweder in den gleichen Kontext verbracht oder in einem neuen Kontext einem akustischen Aversionsreiz ausgesetzt, so kommt es zu verlängertem Freezing. Dieses „Einfrieren“ der Motorik für kurze Zeit weist auf eine Angstreaktion des Tieres hin, die bei Erkennung/Erinnerung des aversiven Reizes auftritt. Die Dauer dieses Freezing im Vergleich zur Kontrollgruppe wird als Gedächtnisleistung interpretiert. Hier zeigte sich, dass bei 28 Tage alten Mäusen, welche über 21 Tage jeden zweiten Tag mit Hochdosis EPO (5000 I.E./kg Körpergewicht) behandelt werden, eine bis zu drei Wochen nach Behandlungsende anhaltende, signifikante Verbesserung der Gedächtnisleistung erzielt wird (Abb. 5).
 bei gesunden, jungen Mäusen. Gezeigt sind Ergebnisse im Fear Conditioning, durchgeführt 1, 3 oder 4 Wochen nach 3wöchiger Hochdosis-EPO-Behandlung. Amygdala-assoziierte Lernprozesse (Cue) werden in dieser Untersuchungsreihe nicht signifikant verändert. Rote Säulen: EPO. Weiße Säulen: Placebo.")
In Zusammenarbeit mit Dr. Weiqi Zhang, Abteilung für Neurophysiologie der Universität Göttingen, untersuchten die Max-Planck-Wissenschaftler zum Zeitpunkt der maximalen Gedächtnisleistung Schnitte lebenden Hippocampusgewebes und konnten zeigen, dass ein typischer Parameter für Lernen und Gedächtnis, nämlich die Langzeitpotenzierung hippocampaler Neurone, durch EPO-Behandlung hochsignifikant gegenüber Kontrolltieren verbessert wird. Gleichzeitig kommt es zu einer Verbesserung der Kurzzeitplastizität und einer Modulierung der synaptischen Übertragung mit Verlagerung des Gleichgewichts von erregender zu hemmender Aktivität (Abb. 6) [12].
 sowie Kurz- und Langzeitpotenzierung (Short-term and long-term potentiation). Vor allem letztere wird als grundlegendes Paradigma für Lern- und Gedächtnisprozesse betrachtet.")
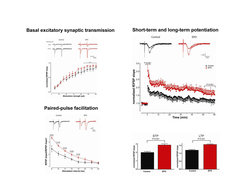
All diese Veränderungen sind unabhängig von den hämatopoietischen EPO-Wirkungen. Es handelt sich um direkte Effekte auf Nervenzellen im Gehirn. Bei kultivierten hippocampalen Neuronen nämlich, welche auf Multi-Elektrode Arrays wachsen, also der Ableitung von sich spontan entwickelnder elektrischer Aktivität zugänglich sind, nimmt EPO direkten Einfluss auf die im Netzwerk entstehenden Entladungen. So scheint EPO bei den sich in vitro entwickelnden neuronalen Netzen zu einer Reduktion von Hintergrundaktivität (Rauschen) beizutragen, wohingegen die Spiking Activity, d. h. das Entladungspotenzial selektiver neuronaler Verbindungen verstärkt wird. Auch auf dem Niveau sich entwickelnder einzelner Nervenzellen im autaptischen Modell (d. h. Nervenzellen in Kultur, die mit sich selbst synaptische Kontakte machen) reduziert EPO den typischen Anstieg von erregender synaptischer Übertragung, ohne dabei jedoch die Zahl der Synapsen zu verändern (Kooperation mit Dr. Jeong Seop Rhee, Abteilung für Molekulare Neurobiologie, MPI für Experimentelle Medizin). Aus diesen Beobachtungen ziehen Ehrenreich und ihre Mitarbeiter die Schlussfolgerung, dass sie es unter EPO-Behandlung mit einem prolongierten funktionellen Silencing (Stilllegung) von Synapsen zu tun haben könnten [12].
Insgesamt lassen sich aus den beschriebenen Untersuchungen die Schlüsse ziehen, dass EPO hippocampusabhängiges Gedächtnis und Lernen über Plastizitätsmodulierung und über Veränderung von synaptischer Konnektivität und vor allem von Aktivität in gedächtnisrelevanten neuronalen Netzen beeinflusst. Diese Wirkmechanismen von EPO müssen nun weiter untersucht und für die Behandlung neurologischer und psychiatrischer Krankheiten nutzbar gemacht werden.
Unter Mitarbeit von (alphabetisch):
Bartosz Adamcio, Ahmed El-Kordi, Benjamin Fischer, Heidi Friedrichs, Martin Fungisai Gerchen, Mohammad Ghorbani, Nora Hagemeyer, Kathrin Hannke, Imam Hassouna, Constanze Hilmes, Silvia Kerbstat, Sabrina Klaus, Richard Leppert, Nina Mertens, Konstantin Radyushkin, Katja Ribbe, Anja Ronnenberg, Derya Sargin, Felix Schellenberger, Julia Sowislo, Kira Späte, Swetlana Sperling, Sabina Stawicki, Nike Stender, Maren Stödtke, Christoph Szuszies, Wiebke Timner, Thilo Wagner, Martin Wegrzyn, Anne Wiedl, Dorina Winter.