Experimental Therapy in Charcot-Marie-Tooth disease
Charcot-Marie-Tooth disease type 1A (CMT1A) is the most common hereditary neuropathy in humans. CMT1A is associated with a DNA duplication on chromosome 17p11.2-p12 that causes an increased gene dosage of peripheral myelin protein 22 (PMP22). Transgenic ("CMT") rats expressing additional copies of this gene show typical characteristics of the human disease such as muscle weakness, reduced nerve conduction velocity, peripheral demyelination and the typical "onion bulb formation".
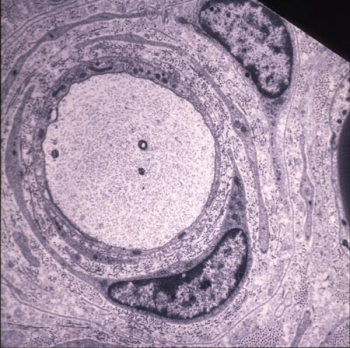 |
|
Typical onion bulb of a 6 month-old „CMT“ rat showing concentric layers of Schwann cell processes and redundant basal laminae around a naked axon. Endoneurial collagen is markedly increased (Sereda et al., Neuron. 1996). |
Targeting myelin lipid metabolism as an effective therapeutic strategy in a rodent model of CMT1A neuropathy
In patients with Charcot-Marie-Tooth disease 1A (CMT1A), peripheral nerves display aberrant myelination during postnatal development, followed by slowly progressive demyelination and axonal loss during adult life. In this experimental approach, we show that myelinating Schwann cells in a rat model of CMT1A exhibit a developmental defect that includes reduced transcription of genes required for myelin lipid biosynthesis. Consequently, lipid incorporation into myelin is reduced, leading to an overall distorted stoichiometry of myelin proteins and lipids with ultrastructural changes of the myelin sheath. Substitution of phosphatidylcholine and phosphatidylethanolamine in the diet is sufficient to overcome the myelination deficit of affected Schwann cells in vivo. This treatment rescues the number of myelinated axons in the peripheral nerves of the CMT rats and leads to a marked amelioration of neuropathic symptoms. We propose that lipid supplementation is an easily translatable potential therapeutic approach in CMT1A and possibly other dysmyelinating neuropathies. (Fledrich et al., Nat Commun. 2018)
 By RNAseq analysis, sciatic nerves from CMT1A (Tg) rats at E21, P6 and P18 revealed a failure in the upregulation of lipid biosynthesis associated genes during primary myelination (Pattern 1, highlighted in magenta). (B) Myelinating DRG Schwann cell cocultures, supplemented with fluorescently labelled phosphatidyl choline (BODIPY-PC, top: green) or phosphatidic acid (BODIPY-L, bottom) show uptake of phosphatidylcholine into the myelin sheath (MBP, magenta). Cell nuclei are counterstained with DAPI (blue). (C) Myelination in DRG neuron Schwann cell cocultures is improved after phosphatidyl choline (PC) supplementation. (D, E) Treatment of CMT rats with a chow enriched in phospholipids (3% PL) resulted in improved myelination. From: Fledrich et al., 2018.")

Fig 1: Perturbed lipid synthesis in CMT1A Schwann cells and therapeutic effect of dietary lipids. (A) By RNAseq analysis, sciatic nerves from CMT1A (Tg) rats at E21, P6 and P18 revealed a failure in the upregulation of lipid biosynthesis associated genes during primary myelination (Pattern 1, highlighted in magenta). (B) Myelinating DRG Schwann cell cocultures, supplemented with fluorescently labelled phosphatidyl choline (BODIPY-PC, top: green) or phosphatidic acid (BODIPY-L, bottom) show uptake of phosphatidylcholine into the myelin sheath (MBP, magenta). Cell nuclei are counterstained with DAPI (blue). (C) Myelination in DRG neuron Schwann cell cocultures is improved after phosphatidyl choline (PC) supplementation. (D, E) Treatment of CMT rats with a chow enriched in phospholipids (3% PL) resulted in improved myelination. From: Fledrich et al., 2018.